
Myasthenia gravis
Die generalisierte Myasthenia gravis (gMG) ist eine seltene chronische Autoimmunkrankheit. Dabei kommt es infolge fehlgesteuerter Immunreaktionen zu Störungen der neuromuskulären Übertragung.
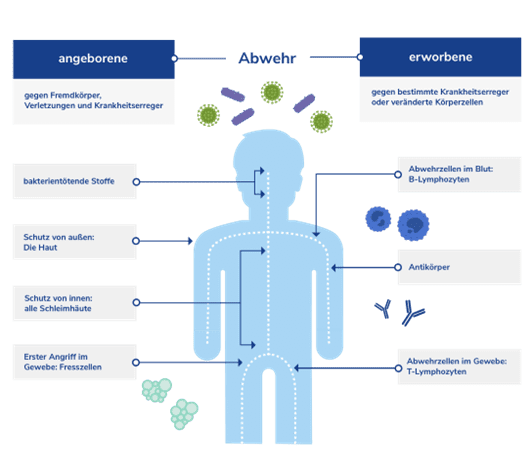
Die generalisierte Myasthenia gravis (gMG) ist eine seltene chronische Autoimmunkrankheit. Dabei kommt es infolge fehlgesteuerter Immunreaktionen zu Störungen der neuromuskulären Übertragung.

Beim atypischen Hämolytisch-Urämischen Syndrom, kurz aHUS genannt, handelt es sich um eine chronische Erkrankung, die durch eine übermäßige Aktivität des Komplementsystems hervorgerufen wird.

Die Hypophosphatasie ist eine erblich bedingte Stoffwechselerkrankung, die sich auf Knochen, Zähne, Muskulatur und weitere Körperfunktionen auswirken kann.

Charakteristisch für die Krankheiten sind Entzündungen von Sehnerv, Rückenmark oder Gehirn, die schubweise auftreten.

Eine komplexe Bluterkrankung mit vielfältigen Symptomen. Hier finden Sie alle Infos.
Die generalisierte Myasthenia gravis (gMG) ist eine seltene chronische Autoimmunkrankheit. Dabei kommt es infolge fehlgesteuerter
Beim atypischen Hämolytisch-Urämischen Syndrom, kurz aHUS genannt, handelt es sich um eine chronische Erkrankung, die
Die Hypophosphatasie ist eine erblich bedingte Stoffwechselerkrankung, die sich auf Knochen, Zähne, Muskulatur und weitere
Charakteristisch für die Krankheiten sind Entzündungen von Sehnerv, Rückenmark oder Gehirn, die schubweise auftreten.
Eine komplexe Bluterkrankung mit vielfältigen Symptomen. Hier finden Sie alle Infos.
Sie sehen gerade einen Platzhalterinhalt von Vimeo. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von YouTube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von hCaptcha laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von Turnstile laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr Informationen


