
Neurofibromatose Typ 1
Neurofibromatosen gehören zu den genetischen Erkrankungen, die sich vor allem in Symptomen, die die Haut und das Nervensystem betreffen, manifestieren.
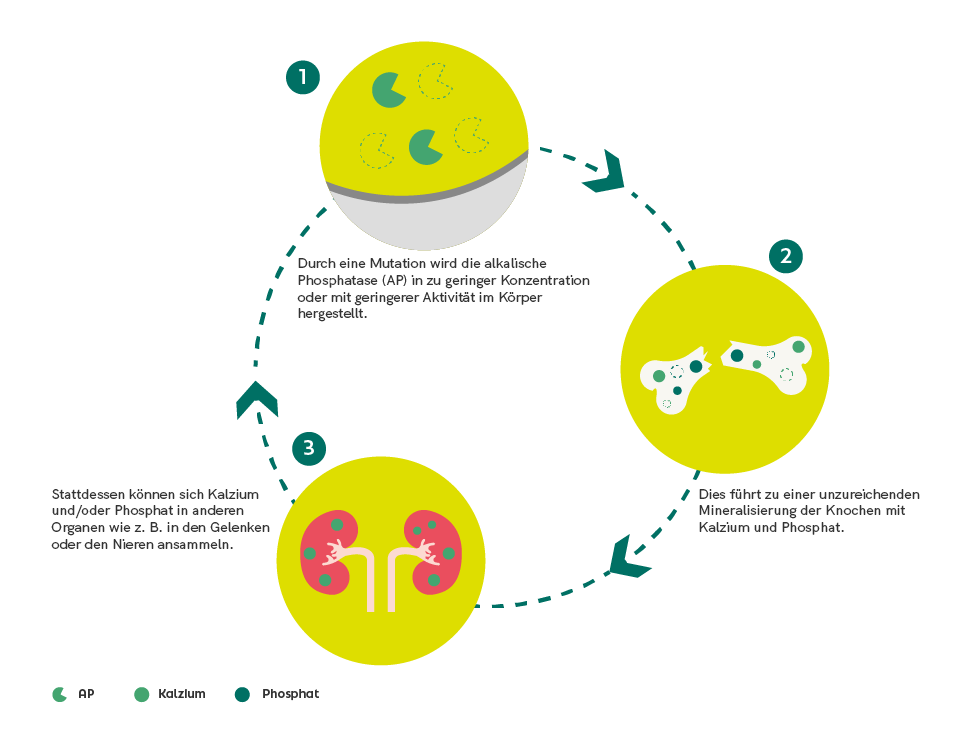
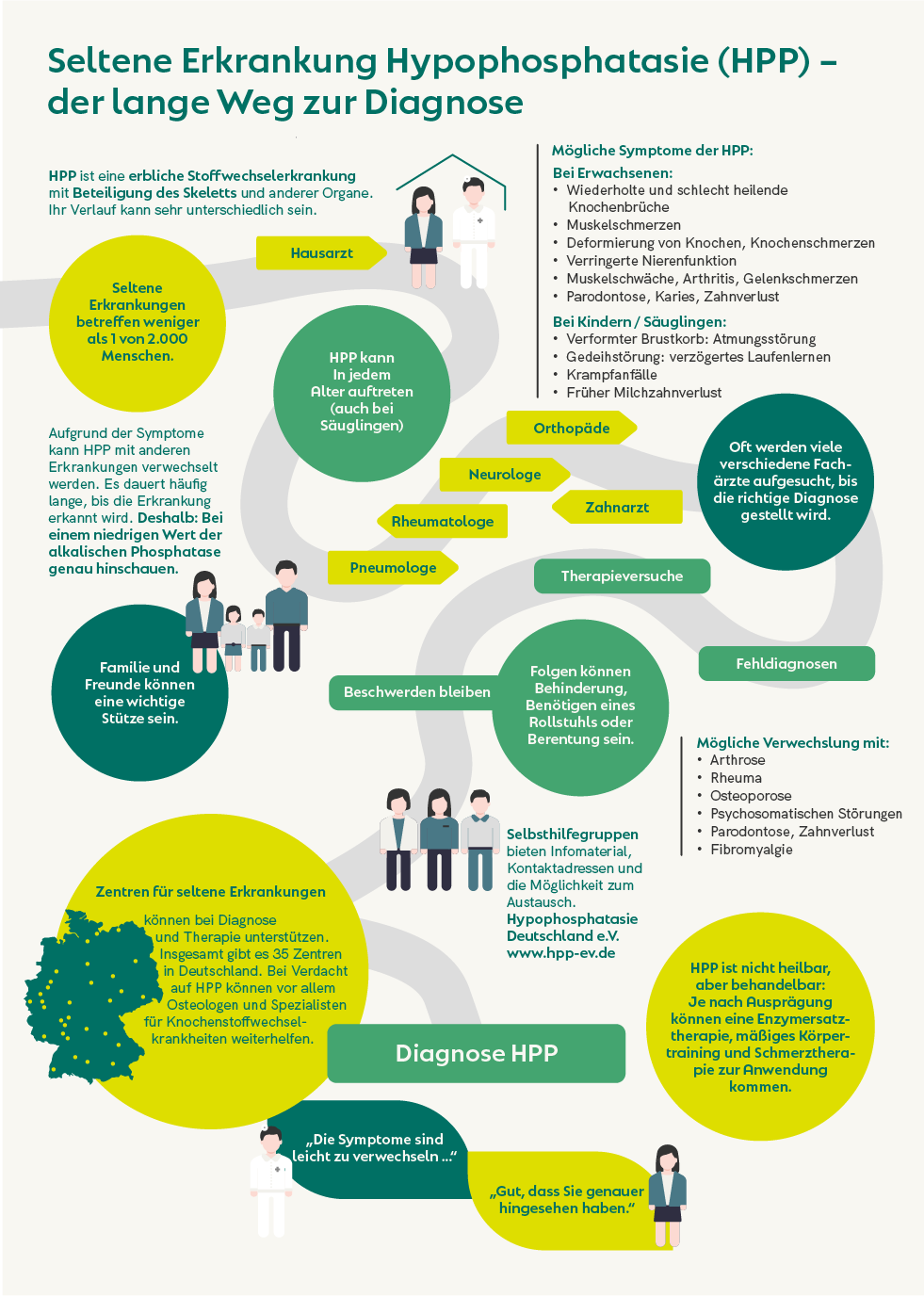
Neurofibromatosen gehören zu den genetischen Erkrankungen, die sich vor allem in Symptomen, die die Haut und das Nervensystem betreffen, manifestieren.

Die generalisierte Myasthenia gravis (gMG) ist eine seltene chronische Autoimmunkrankheit. Dabei kommt es infolge fehlgesteuerter Immunreaktionen zu Störungen der neuromuskulären Übertragung.

Morbus Wilson ist eine seltene vererbbare Stoffwechselerkrankung. Infolge einer genetischen Störung kann Kupfer nicht richtig ausgeschieden werden und lagert sich stattdessen in verschiedenen Organen an.

Erfahre mehr über die Hyposensibilisierung, eine effektive Immuntherapie zur Behandlung von Allergien.

Erfahre mehr über Wasserallergie und ihre Symptome. Eine Wasserallergie, auch bekannt als aquagene Urtikaria, ist eine seltene Form von allergischer Reaktion, bei der Menschen empfindlich auf Wasser reagieren.
Sie sehen gerade einen Platzhalterinhalt von Vimeo. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von YouTube. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie sehen gerade einen Platzhalterinhalt von Facebook. Um auf den eigentlichen Inhalt zuzugreifen, klicken Sie auf die Schaltfläche unten. Bitte beachten Sie, dass dabei Daten an Drittanbieter weitergegeben werden.
Mehr InformationenSie müssen den Inhalt von hCaptcha - Formidable laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von reCAPTCHA laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr InformationenSie müssen den Inhalt von Turnstile laden, um das Formular abzuschicken. Bitte beachten Sie, dass dabei Daten mit Drittanbietern ausgetauscht werden.
Mehr Informationen

